EA Spectroscopy as a series of sensors: Investigating the Impact of Solvent Type on Mobility in Organic Diodes
Team members
Li Jinhan A0327554Y
Liu Chenyang A0328377R
Idea
We will use EA spectroscopy, which will include optical sensors, electrical sensors, and lock-in amplifiers, among other components as a highly sensitive, non-destructive optical sensing platform to measure the internal electric field modulation response of organic diodes under operating conditions, and to quantitatively extract carrier mobility based on this measurement. By systematically controlling the thin film preparation temperature and comparing the EA response characteristics of different samples, the project aims to reveal the influence of film preparation temperature on device mobility.
Introduction
The performance of organic semiconductor devices (such as organic diodes) is largely limited by the charge transport processes within the thin film, and carrier mobility is one of the key parameters characterizing charge transport capability. Since organic thin films typically exhibit significant morphological and microstructure sensitivity, the film fabrication temperature affects factors such as molecular packing, crystallinity, phase separation behavior, and trapped state density, thereby altering the internal electric field distribution and charge injection/transport efficiency, ultimately manifesting as differences in mobility and device response. Therefore, establishing a characterization method capable of reliably tracking the "process-structure-transport" relationship is crucial for process optimization and device performance improvement.
EA Theory
Electroabsorption technology is achieved by measuring the change in absorption coefficient after an applied electric field is applied. Under normal incident conditions, the intensity of light transmitted through the absorbing medium can be calculated using the Lambert-Beer law:
Where represents the original light intensity of the incident light, is the reflectivity, is the absorption coefficient, and is the thickness of the absorbing medium.
Both and are affected by the external electric field, and their changes as follows:
Under classical operating conditions, the change in the value of is negligible. Equation (2) simplifies to:
From a microscopic perspective, the energy level E(F) of state in electric field F is given by the following equation:
represents the electric dipole moment of this state, and is its polarizability. Therefore, the optical transition energy shift from the initial state to the final state is given by the following equation:
The first term represents the linear energy change caused by the difference in dipole moments between the initial and final states, which cancels out in isotropic solids without permanent dipoles. The second term describes the energy shift caused by the difference in polarizability between the initial and final states, an effect that always exists.
The absorption variation of the external electric field can be represented by the term of the Maclaurin series, while the third and higher order terms can be ignored due to the small spectral changes.
The sole contribution of the first term comes from the second-order Strac effect , while the isotropic average of in the second term does not cancel out, but instead produces in the randomly distributed matrix. The third contribution of comes from the transition of the oscillator to the previously forbidden state when the electric field is present, and this transition also has a quadratic relationship with F.
Adding up all contributions to , we get:

For a composite electric field with AC component and DC component :
Substituting formula (8) into (7) yields the modulation of at the fundamental frequencies and :
represents the internal electric field generated by the equilibrium state of the two electrodes EF.
A phase-sensitive lock-in amplifier can measure the component:
and components:
Since and are linearly related, the internal field can be determined by measuring required to eliminate the electroabsorption response at .
EA device setup and operation procedures
A schematic diagram and photograph of the homemade electro absorption device are shown in the figure.
This device is driven by a modulated driving voltage. The DC bias voltage varies from 0V to 3V in 0.5V steps to change the electric field strength, while a constant AC bias voltage is applied simultaneously. The change in electric field strength after applying this driving voltage leads to a change in the absorption bandgap .
Monochromatic light is incident on a glass substrate at a 45° angle, and the reflected light illuminates a photodiode.
The 1\omega component is proportional to , and can therefore be used to determine the built-in potential of the device. In this experiment, phase-sensitive lock-in detection was employed to extract the first harmonic component of the EA signal, , and thereby determine . An AC modulation field, , was superimposed on the DC bias , causing the reflection signal to vary periodically with time and contain multiple harmonic components, such as and .
The signal detected by the sensor was subsequently fed into a lock-in amplifier. Using the applied AC modulation signal as the reference, the lock-in amplifier selectively extracts the signal component at the reference frequency, , and with a fixed phase relationship by multiplying the input signal with the reference signal followed by low-pass filtering. This process demodulates the original AC signal into a DC output, whose magnitude is proportional to the intensity of the component.
Therefore, by locking to the frequency, the first harmonic component of the EA signal can be effectively isolated and measured, enabling quantitative analysis of the built-in electric field.

Experimental Principle
Built-in potential is the potential difference formed by the internal charge distribution of a material when no external voltage is applied.
Under the space charge confined current (SCLC) mechanism, the relationship between current density and carrier mobility follows the Mott-Gurney law:
Where is the vacuum permittivity, is the relative permittivity of the material, is the built-in potential, and is the thickness of the organic layer. This relationship shows that the current density is proportional to the carrier mobility , and also strongly depends on the built-in potential and the film thickness .
Mobility is obtained using the Mott-Gurney law. The J-V relationship in the formula can be directly measured, but is missing. Therefore, the Accurate Estimation Method (EA) is used to obtain the accurate for calculating mobility. The working principle of the EA has been explained in detail above. The following section explains how to use the EA to obtain and how to derive the carrier mobility .
How to get :
Bulit-in potential is , Applied Voltage is . Internal electric field :
Because the device is made of Ag and ITO, the absorption intensity is difficult to detect. The detector detects the reflected light, and since the intensity of the reflected light is very weak, we use phase-locked detection to output to enhance the signal strength.
Because:
So:
Then:
Using trigonometric formulas:
The resulting absorption change is as follows:
For empty current:
As shown in the figure below, by changing the input value in the data, when the null current curve flips, the voltage value at the flip point is .

Derivation of the calculation of carrier concentration:
By performing EA, we can get of the devices. By applying the equations below, we can calculate carrier mobility.
Gauss’s Law: , in 1D: .
Drift-diffusion equation: .
MG equation: .
Combining the above three formulas yields:
Since we assume the electric field across OSC is constant, we can ignore the term, and:
From Gauss’s Law, we can calculate the carrier density:
The average carrier density is:
In Mott-Gurney regime:
The final result is:
How solvent effect aggregation
For polymer systems, solvents are generally classified as either good solvents or poor solvents. In a good solvent, strong polymer–solvent interactions enable effective solvation of the polymer chains, such that the chains adopt relatively extended conformations and remain well dispersed, with only a weak tendency toward aggregation. By contrast, in a poor solvent, polymer–solvent interactions are comparatively weak, while interchain interactions become more significant. Under these conditions, the polymer chains tend to contract, entangle, and aggregate, and may eventually precipitate from solution.
For conjugated polymers, however, this binary classification is often insufficient to describe their solution behavior in a comprehensive manner. Zhao et al. introduced the concept of a borderline solvent, defined as a solvent with a solvating ability intermediate between that of a good solvent and a poor solvent. In such a medium, the polymer chains are not fully extended and uniformly dispersed, as would be expected in a good solvent. Instead, they are more prone to intermolecular approach and the formation of a certain degree of ordered aggregation, while remaining dispersed in solution without obvious precipitation. Borderline solvents therefore have particular significance in the regulation of aggregation in conjugated polymer systems, and consequently exert an important influence on subsequent film formation, thin-film morphology, and device performance.
As shown in Figure 4, Zhao et al. proposed a mechanism by which a borderline solvent can induce ordered aggregation in conjugated polymers. In a borderline solvent, the polymer chains are more likely to undergo an early transition from coil-like conformations to more extended chain conformations, and subsequently form small, ordered 𝜋-stacking aggregates. These initial aggregates can then further organize and grow into more distinct plate-like stacked structures with a high degree of order and a preferred orientation. During the subsequent spin-coating process, the aggregates continue to spread, develop, and align on the substrate, thereby promoting the formation of an ordered thin film. Zhao et al. suggested that such dilute-solution 𝜋-stacking and the resulting extended-chain aggregates are crucial for achieving pronounced lamellar order in conjugated polymer films.
By contrast, in a good solvent, polymer–solvent interactions are sufficiently strong to maintain the polymer chains in a more fully solvated and dispersed state. Under these conditions, significant 𝜋-stacking tends to occur only at a later stage of film formation. At that point, however, the polymer chains have already become more entangled, and the adjacent 𝜋-stacked aggregates are more likely to interpenetrate and entangle with one another. This suppresses the formation of extended-chain 𝜋-stacks and reduces the degree of structural order in the resulting film. Consequently, films processed from a good solvent typically contain a larger fraction of disordered or amorphous regions.
These observations demonstrate that the solvent plays a decisive role in regulating polymer aggregation behavior. It influences not only the conformation and dispersion state of the polymer chains in solution, but also the onset, extent, and degree of order of aggregation during film formation. Therefore, solvent selection is a key factor in controlling polymer aggregation, and it can subsequently affect thin-film morphology and charge-transport properties.

Result of EA
From the theory of EA, Vbi of different device can be measured. Here is the result of EA:
-
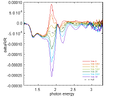 400px
400px -
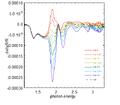 400px
400px -
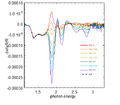 400px
400px



Result of carrier mobility
Based on the above equations, the carrier mobility was calculated using the J–V curve data, and the results are shown below.
Conclusion
By cross-comparing the charge carrier mobility of devices fabricated with different film thicknesses, for both PBDB-T and PM6, CB is a more suitable solvent than CF. In the CB system, both polymers show better solubility and stronger aggregation in solution, and the corresponding films show higher carrier mobility. In addition, the CB-based results are consistent under different solution conditions: the behavior observed in the 120 nm films can be viewed as an extension of the 180 nm current–carrier density (CCD) results, while solution heating has only a small effect on mobility. These results suggest that, within the processing range studied here, the charge-transport behavior remains relatively stable, and the film morphology is still in a range that is favorable for carrier transport.
Based on these results, it can be suggested that, within a proper range, stronger aggregation is related to higher carrier mobility. It can be inferred from this that, there seems to be an aggregation window, in which increased aggregation helps improve charge transport. This idea is consistent with the aggregation theory proposed by Zhao et al., which suggests that moderate aggregation can improve molecular packing, strengthen interchain coupling, and form more continuous charge-transport pathways.
A possible explanation is that moderate aggregation provides a certain degree of molecular pre-organization before or during film formation. This pre-organization may help the film form more ordered molecular packing and better-connected transport pathways, which is beneficial for carrier transport. However, this positive effect only exists within an intermediate range. When aggregation is too weak, molecular pre-organization is not enough, so π–π stacking forms too late during film formation, which may lead to chain entanglement, lower order, and poor pathway connectivity. In contrast, when aggregation becomes too strong, the polymer may form overly large domains, fibers, or particles too early, leading to morphology that is less uniform, weaker connection between domains, and more defects or trap effects. As a result, once the aggregation strength goes beyond the proper window, the benefit from better local order may be offset by the loss of film uniformity and long-range transport continuity, and the mobility may decrease.